
Sie befinden sich hier: Home : Industriegase Lexikon: Medizinische Gase : Medizinische Druckluft
Medizinische Druckluft ist ein Arzneimittel. Demzufolge gelten für die Erzeugung dieser medizinischen Druckluft auch einzuhaltende Verordnungen, Richtlinien, Normen und Gesetzte sowie festgelegte Grenzwerte.
Die Grenzwerte für die medizinische Druckluft werden im Europäischen Arzneimittelbuch geregelt. Unter der Bezeichnung "Aer medicinalis" ist dort die Monographie hinterlegt.
| Gas | Chemisches Zeichen | Grenzwert |
| Kohlendioxid | CO2 | ≤500 ppm |
| Kohlenmonoxid | CO | ≤5 ppm |
| Schwefeldioxid | SO2 | ≤1 ppm |
| Stickstoffmonoxid | NO | ≤2 ppm |
| Stickstoffdioxid | NO2 | ≤2 ppm |
| Wasserdampfgehalt | H2O | ≤67 ppm / ≤870 ppm* |
| Sauerstoff | O2 | 20,4 % - 21,4 % |
| Ölgehalt | ≤0,1 mg/m3 | |
| Feststoffe | Keine Angaben |
| Gas | Chemisches Zeichen |
Grenzwert |
| Kohlendioxid | CO2 | ≤500 ppm |
| Kohlenmonoxid | CO | ≤5 ppm |
| Schwefeldioxid | SO2 | ≤1 ppm |
| Stickstoffmonoxid | NO | ≤2 ppm |
| Stickstoffdioxid | NO2 | ≤2 ppm |
| Wasserdampfgehalt | H2O | ≤67 ppm / ≤870 ppm* |
| Sauerstoff | O2 | 20,4 % - 21,4 % |
| Ölgehalt | ≤0,1 mg/m3 | |
| Feststoffe | Keine Angaben |
*Mit Ausnahmegenehmigung (siehe weiter unten Ausnahmeregelung)
Die Pharmacopoea Europaea (Ph. Eur.) ist weiterhin bekannt als:
Die Meßmethoden zum Nachweis der Verunreinigungen werden in der Pharmacopoea Europaea / European Pharmacopeia (Ph. Eur.) ebenfalls geregelt. So ist es z. B. nur zulässig, den Wirkstoff (also den Sauerstoff) mit einem Sauerstoff-Meßgerät zu messen, was auf dem paramagnetischen Meßprinzip beruht. Andere Verfahren sind nur zulässig, wenn diese validiert wurden (weitere Informationen erhalten Sie über das BfArM).
Für die Herstellung eines Arzneimittels bedarfs es einer Herstellungserlaubnis. Für Krankenhäuser gibt es gemäß Arzneimittelgesetz eine Ausnahe, sofern diese die Kriterien erfüllen.
Diese Ausnahme wird über das Arzneimittelgestzt (AMG) geregelt.
(1) Wer 1. Arzneimittel im Sinne des § 2 Absatz 1 oder Absatz 2 Nummer 1,
gewerbs- oder berufsmäßig herstellt, bedarf einer Erlaubnis der zuständigen Behörde.
(2) Einer Erlaubnis nach Absatz 1 bedarf nicht 1.
1. (……)
2. der Träger eines Krankenhauses, soweit er nach dem Gesetz über das Apothekenwesen Arzneimittel abgeben darf, oder für die Rekonstitution oder das Abpacken einschließlich der Kennzeichnung von Arzneimitteln, die zur klinischen Prüfung bestimmt sind, sofern dies dem Prüfplan entspricht,
Die Freigabe sowie die fortlaufende Prüfung des Arzneimittels "medizinische Druckluft" bzw. "Aer medicinalis" wird über die Apothekenbetriebsordnung - ApBetrO geregelt.
Der Apotheker sollte eine entsprechende SOP erstellt haben, in der alle relevanten Punkte zur erstmaligen Freigabe der Produktionsanlage, Freigabe des Arzneimittels, wiederkehrende Prüfungen des Arzneimittels sowie die Verantwortlichkeiten geregelt werden. Bei der wiederkehrenden Prüfung des Arzneimittels Aer medicinalis ist unbedingt auf die Einhaltung der in der Pharmacopeia (Ph. Eur.) genannten Grenzwerte und deren Messmethode zu achten.
Wenn z. B. aus Kostengründen auf die Messung des Wirkstoffs "Sauerstoff" verzichtet wird, oder ein nicht validiertes Verfahren angewendet wird, kann diese Qualitätsmessung nicht zur Freigabe/Prüfung der medizinischen Druckluft / Aer medicinalis herangezogen werden.
Bei Anlagen zur Herstellung bzw. Erzeugung medizinischer Druckluft / Aer medicinalis handelt es sich um Medizinprodukte.
Hersteller, die ein Medizinprodukt herstellen, müssen gemäß 93/42 EWG zertifiziert sein und ein Qualitätsmanagement System entsprechend DIN EN ISO 13485 aufrecht erhalten bzw. von einer benannten Stelle zertifiziert worden sein.
Medizinprodukte werden über das Medizinproduktegesetzt (MPG) geregelt.
Zweck dieses Gesetzes ist es, den Verkehr mit Medizinprodukten zu regeln und dadurch für die Sicherheit, Eignung und Leistung der Medizinprodukte sowie die Gesundheit und den erforderlichen Schutz der Patienten, Anwender und Dritter zu sorgen.
Zu beachten ist hierbei, dass das MPG abweichend vom AMG nicht nur den Schutz der Patienten in den Vordergrund stellt sondern auch Anwender und Dritte einbezieht.
Das Medizinprodukt setzt sich zusammen aus dem
Medizinprodukt = Produkt + Produktinformationen
Das Produkt setzt sich zusammen aus den Einzelkomponenten sowie der Zusammensetzung, der Errichtung/Installation, Inbetriebnahme, Übergabe/Einweisung
Die Grundlegenden Anforderungen müssen dem Stand der Technik zum Zeitpunkt der Konzeption ensprechen. Der Schutz von Gesundheit und Sicherheit müssen dem MPG Rechnung tragen.
Es müssen Maßnahmen getroffen werden, damit von einem Produkt ausgehende Gefahren abgewendet werden und somit eine Verletzung von Rechtsgütern Dritter ausgeschlossen wird.
Die Errichtung einer Anlage zur Erzeugung und Bereitstellung von medizinischer Druckluft (Aer medicinalis) wird über die DIN EN ISO 7396-1 geregelt. In dieser Norm werden grundlegende Anforderungen sowie
- Montage
- Installation
- Prüfungen
- Dokumentation
behandelt.
Zu den grundlegenden Anforderungen gehört z. B. das "drei Quellen Prinzip". In der neuesten Fassung wird darauf verwiesen, dass die dritte Quelle in einem separaten Raum zu installieren ist.
Unabhängig von den Forderungen der DIN EN ISO 7396-1 gelten selbstverständlich weitere Normen, Richtlinien oder Verordnungen die z. B. mit dem Betrieb von Arbeitsmittel oder Druckgeräten zu tun haben. Zu nennen sind hier exemplarisch die Druckgeräterichtlinie oder die Betriebssicherheitsverordnung (BetrSichV) sowie das Arbeitsschutzgesetz (ArbSchG) usw.
Heutige Anlagen zur Erzeugung medizinischer Druckluft bestehen z. B. aus
Für Hersteller von Medizinprodukten gemäß der Richtlinien für Medizinprodukte (93/42/EWG) wird sich jedoch spätestens im Jahre 2020 eine Verschärfung ergeben. Die Richtlinien für Medizinprodukte (93/42/EWG) und aktive implantierbare Medizinprodukte (90/385/EWG) werden mit einer Übergangsfrist von drei Jahren zu einer Medizinprodukte-Verordnung zusammengeführt und verlieren anschließend ihre Gültigkeit. Ab Mitte 2020 gilt dann nur noch die Medizinprodukte-Verordnung (MDR) und nicht mehr die Medizinprodukte-Richtlinie (MDD).
Durch diese neue Verordnung werden Hersteller stärker verantwortlich gemacht für die Rückverfolgbarkeit ihrer Produkte in Bezug auf Sicherheit, Leistung und Qualität der in Verkehr gebrachten Medizinprodukte.
Im Rahmen der Novellierung der MEDDEV 2.7/1 rev. 4 ist seit Juli 2016 für alle Hersteller von Medizinprodukten eine klinische Bewertung notwendig geworden (Siehe hierzu MEDDEV 2.7/1 Kapitel 11. The clinical evaluation report (CER, Stage 4)). Zertifizierte Unternehmen können Ihnen auf Verlangen ihre klinische Bewertung für ihre Produkte vorlegen.
Lassen Sie sich doch einfach mal die Bewertung zeigen.
Im Jahr 2007 entschied die ZLG eine Ausnahmereglung für den Feuchtegehalt von im Krankenhaus erzeugter medizinischer Druckluft zuzulassen (siehe hierzu ZLG EFG Votum V15004). Im Jahr 2008 wurde durch die ZLG ein Dokument verifiziert zur Vereinheitlichung der Inspektionsanforderungen und der Vorgehensweise bei Inspektionen in Herstellungsbetrieben für medizinische Gase (siehe hierzu Aide-memoire 07121401). Die Ausnahmeregelung wurde ebenfalls in die Monograpie für Aer medicinalis im Arzneimbuch aufgenommen.
"Für Luft sind im EuAB anlagenabhängig unterschiedliche Grenzwerte für den
Wassergehalt spezifiziert. So kann dieser maximal 870 ppm betragen, wenn das
Versorgungssystem für Druckluft einen Druck von maximal 10 bar aufweist und
die Rohrleitungen nicht in Bereichen verlegt sind, in denen Temperaturen unterhalb
von 5 °C auftreten. Ansonsten sind 67 ppm vorgeschrieben.
Hieraus ergibt sich, dass unterschiedliche Konzepte der Trocknung des Gases
nach der Kompression möglich sind. Dabei werden derzeit in den Erzeugungsanlagen
Kälte- und Adsorptionstrockner zur Sicherstellung der notwendigen
Reduzierung des Wassergehaltes eingesetzt.
Im Fall der Trocknung mittels Kältetrocknern (Drucktaupunkt etwa 3 °C) ist nicht
immer sichergestellt, dass die Spezifikation des EuAB für den Wassergehalt
eingehalten wird. Eine ständige Messung der Feuchte mit entsprechender Alarmierung bei Grenzwertüberschreitung sollte sichergestellt sein.
Im Fall der Installation von Adsorptionstrocknern soll durch das Arbeitsprinzip
der Trockner und die meist integrierte Taupunktüberwachung ein Feuchtegehalt
unterhalb von 67 ppm sichergestellt werden."
Auf technische Details wird allerdings nicht eingegangen. Was passiert z. B. wenn nur ein Kältetrockner installiert ist und durch Vereisung eine Blockade die Druckluftbereitstellung unterbindet. Was passiert, wenn nur ein Trockner installiert ist und dieser ausfällt? Wie wird in beiden Fällen eine sichere Versorgung gewährleiste?
Welcher Druck ist gemeint und was ist mit Versorgungssystem gemeint? Vermutlich ist mit 10 bar der Absolutdruck gemeint, da dies zu 870 ppm Feuchtegehalt bei 5°C führen würde (Absolutdruck N.N. 1,01325 bar ergibt bei 9 bar ü total 10,01325 bar abs. und bei 5°C Taupunkt einen Feuchtegehalt von 870,36 ppm was nach gängigen Abrundungskriterien dann als 10 bar, 870 ppm geschrieben werden kann). Weiterhin sollte der Druck im Puffer gemeint sein.
| Druck [abs] | Taupunkt | Feuchte |
| 9 | 5°C | 968 ppm |
| 10 | 5°C | 871 ppm |
| 10,01325 | 5°C | 870,36 ppm |
| 14 | 5°C | 623 ppm |
Das Diagramm unterhalb zeigt den Feuchtegehalt bei 5°C und dem im Diagramm dargestellten Druck. Jeglicher Druck unterhalb von 10 bar abs. (9 bar ü) führt zu einem unzulässig hohen Feuchtegehalt der medizinischen Druckluft / Aer medicinalis
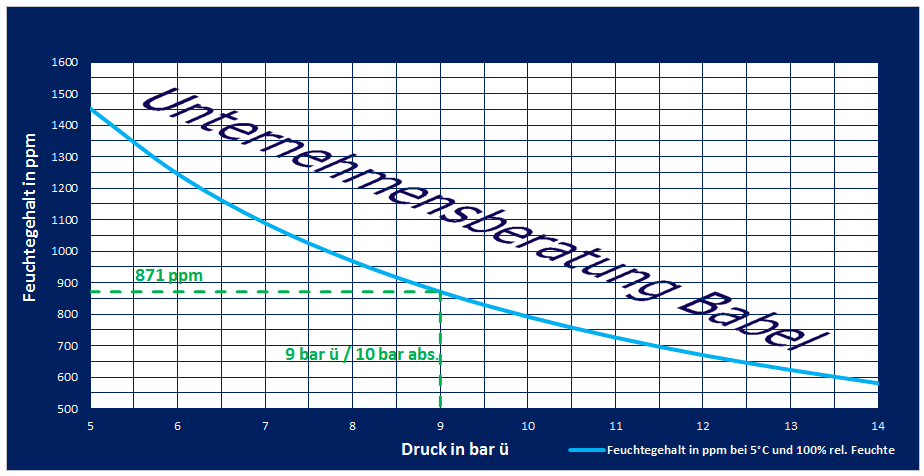
Logisch wäre somit eine Formulierung gewesen, die einen Mindestdruck von 10 bar absolut hinter dem Kompressor bzw. im Puffer vorschreibt. Viele Systeme lassen allerdings einen unteren Pufferdruck von 9 bar abs. bzw. 8 bar Überdruck oder weniger zu. Eine Kontrolle des Feuchtegehaltes findet dann aber nicht mehr statt, da sich die Betreiber auf den eingestellten Taupunkt am Kältetrockner verlassen.
Dies ist bei geringeren Drücken eine fatale Fehleinschätzung.
Die unglückliche Formulierung in der ZLG Freistellung kann dazu führen, dass ein Betreiber seine Anlageneinstellung so wählt, dass ein Grenzwert von 10 bar nicht überschritten wird. Dies hat ebenfalls zur Folge, dass der maximal zulässige Höchstwert von Wasserdampf (≤870 ppm) regelmäßig überschritten wird. Leider hat es nie eine Klarstellung gegeben und der Betreiber sollte über das nötige Know how verfügen, die richtigen Schlüsse zu ziehen.
Durch die Novellierung der MEDDEV 2.7/1 sind die Hersteller verpflichtet, eine klinische Bewertung durchzuführen. Diese Bewertung regelt die Einsatzbedingungen bzw. die Rahmenbedingungen, unter denen die medizinische Druckluft hergestellt wird. Es ist darauf zu achten, dass in der klinischen Bewertung Bezug auf den Feuchtegehalt genommen wird. Sofern dies gemacht wurde stellt sich die Frage, ob gemäß klinischer Bewertung ein Feuchtegehalt oberhalb von 67 ppm überhaupt zulässig ist.


Medizinische Druckluft gehört quasi zur Grundausstattung eines jeden Krankenhauses.

Eine Fehlplanung kann fatale Folgen haben.
